The mechanisms of sex determination in animals are remarkably diverse. Gonochoristic animals show genetic and/ or environmental sex-determining mechanisms. Genetic sex-determining systems can be either chromosomal, and involve a master sex-determining gene/region on a sex chromosome, or can be polygenic and involve several genes/ regions on multiple chromosomes.
In most fish species with XX/XY or ZZ/ZW mechanism, the sex chromosomes do not show clear differences in length or gene content. Several fish sex determining genes have been isolated from species with XX/XY mechanisms: DMY/dmrt1bY in Oryzias latipes (medaka); Gsdf(Y) in Oryzias luzonensis (Luzon ricefish); amhy in Odontesthes hatcheri (Patagonian pejerrey); Amhr2 in Takifugu rubripes (tiger pufferfish); and sdY in Oncorhynchus mykiss (rainbow trout).
In environmental sex-determining systems, the environment plays a decisive role, such as temperature in turtles, alligators and fish. Both systems can interact in some species such as in O. latipes, which has an XX/XY genetic system, where high temperatures can cause female-tomale sex reversal. Additionally, autosomal loci can also contribute to sex determination in many species.
Overall, the understanding of sex determination systems in fish has direct commercial applications, given the strong sexual dimorphism exhibited in a wide variety of aquaculture fish species for a range of commercially important traits like growth or age at maturity.
Hippoglossus hippoglossus (Atlantic halibut) has been a high-value species for cold-water marine aquaculture for several decades in Northern Europe and America, although production has been limited by a series of bottlenecks.
Among these, sexual dimorphism in growth, with males maturing earlier and growing significantly slower than females, reduces productivity and profitability of the sector. Females can reach market size (3-5 Kg) at around 36 months while males require at least an extra year, making the production of all-female stocks particularly appealing for the aquaculture industry.
Flatfish (order Pleuronectiformes) show a range of sexdetermining mechanisms, including XX/XY and ZZ/ZW, with significant effects of environmental factors, principally temperature, in some species.
Meiotic gynogenetic H. hippoglossus were all-female, suggesting an XX/ XY sex-determining system. Temperature has not been shown to have an effect on H. hippoglossus sex ratio. Gonadal sex differentiation can be manipulated through in-feed synthetic steroid treatments (e.g., 17 α-methylhydrotestosterone, MDHT or 17 β-estradiol) or aromatase inhibitor treatments (e.g., Fadrozole). However, direct sex reversal is not a commercially acceptable means to alter sex ratios in food fish within the EU. Thus indirect sex reversal is required, whereby masculinised genotypic females (XX neomales) are crossed to normal females (XX) to produce genetically all-female progeny, a process which has yet to be proven in H. hippoglossus. The crux of successful indirect sex reversal is the non-lethal identification of the neomales.
Currently the main technique for such verification is progeny testing of treated animals which is time consuming and costly, taking at least four or five years due to the timing of puberty in halibut (reached after three years). Direct genetic sexing, instead of progeny testing, would be preferable using non-lethal and cheap genotyping techniques. This is only likely to be possible in simple cases of male or female heterogamety. Sex-specific genomic sequences are only available in a limited number of aquaculture species. Although a genetic linkage map based on microsatellites and amplified fragment length polymorphism (AFLP) is available for H. hippoglossus, this does not contain any information about sexdetermination. Restriction associated DNA (RAD) sequencing is a powerful technique for generation of high-density linkage maps and conducting quantitative trait locus (QTL) analysis including the mapping of sex-determining loci in fish.
Results
Hormonal sex reversal and neomale verification
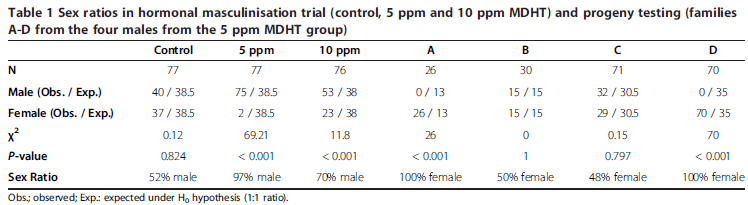
The control group exhibited a sex ratio not significantly different from 1:1 (52 per cent ?; 48 per cent ?), whereas 97 per cent of the group treated for six weeks (5 ppm) and 70 per cent of the group treated for three weeks (10 ppm) were confirmed as phenotypic males (Table 1). Both in-feed treatments significantly altered the natural sex ratio in favour of males. Of the seven putative neomales that were progeny tested, only four crosses produced enough survivors at the age of sexing, at approximately one year of age. From these four crosses, two gave 100 per cent female progeny (Families A and D; Table 1) while the other two gave balanced sex ratios (Families B and C; Table 1).
RAD sequencing
Two crosses with 62 (Family C) and 28 offspring (Family B) and their parents (including a common female) were analysed (Additional file 1). The DNA samples were barcoded, pooled and sequenced in two lanes of an Illuminia HiSeq 2000 sequencer (see Methods). In total, 648,402,546 raw reads (101 bases long) were produced (or 324,201,273 paired-ended reads: NCBI BioProject accession number SRP016043). After removing low quality sequences (quality score under 30), ambiguous barcodes and orphaned paired-end reads, 81.24 per cent of the raw reads were retained (526,783,920 reads). The Stacks package [25] was then used to make a de-novo assembly of the sampled loci from each individual: 82,745 and 83,668 RAD-tags were retrieved for Families B and C respectively, covering 90,105 RAD-tags in total including 76,308 of these shared between the two families (Figure 1). The number of reads and RAD-tags for each sample are reported in Additional file 1.

Genetic map
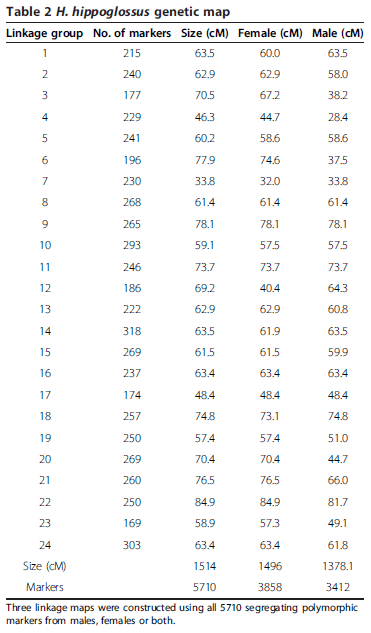
In order to maximise the number of informative markers and minimise the amount of missing or erroneous data, we used only paired-end RAD-tags retrieved in at least 75 per cent of the samples in each family, and carrying one or two SNPs. 7572 and 5954 RAD-tags were retained for the families B, and C respectively (Figure 1). Since Family B had only 28 offspring, the genetic map was constructed with the Family C data only (62 offspring). The map consists of 5703 SNPs and 7 microsatellites (used initially for parentage assignment) in 24 linkage groups (LG) and spanning 1514 cM (Figure 2; Additional file 2). 4049 of the above SNPs were common in the two families and were used to incorporate the data from Family B into a joint linkage map. Sexspecific genetic maps were constructed, with the femalespecific map spanning 1496 cM and the male-specific map spanning 1378 cM (Table 2).
QTL-association mapping
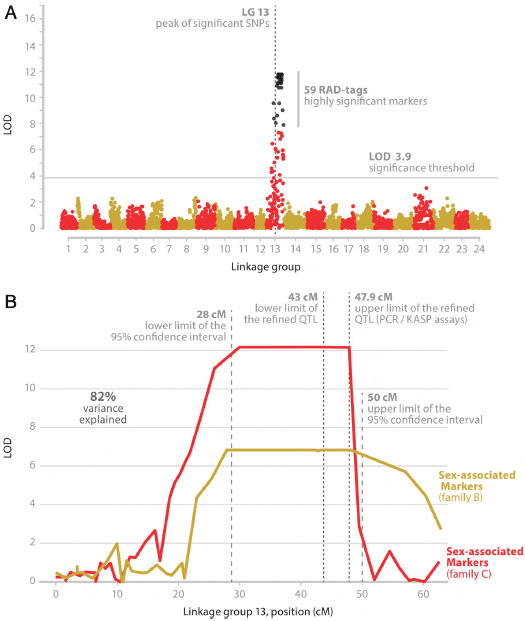
The results from the single-QTL model for binary traits provided evidence for the existence of a major QTL in LG 13 for both families in the male informative dataset (Figure 3A). The highest logarithm of odds (LOD) scores for Families B and C were 6.83 and 12.17 respectively (Figure 3B). The genome-wide thresholds (α = 0.01) were calculated from permutation tests (10,000 permutations) to be 3.25 and 3.90 respectively. The highest LOD scores were observed in the region between 30 cM and 48 cM in LG 13 for both families. Even though models that take into account the existence of a major QTL (as in this study) or ones that test for existence of multiple QTLs simultaneously reduce the residual variation (providing this way higher power in the analysis for detecting additional QTLs at least of modest effect), no additional QTLs were detected. The calculated 95 per cent Bayesian Density Intervals for the QTL location spanned a region of 22 cM (28-50 cM in LG 13), while the 1.5-LOD support interval spanned a region of 28 cM (23-51 cM in LG 13). The variance explained by the QTL was around 82 per cent. There were no significant QTL in the female informative dataset. The Polymorphism Information Content (PIC) of the markers in LG 13 ranged from 0.64 to 0.99. The joint QTL analysis of the two families showed a maximum at 46 cM with a Likelihood Ratio (LR) of 96 (Figure 3B). The chromosome-wide threshold calculated from permutation tests (10,000 permutations) was LR = 19. The association analysis identified 38 SNPs being strongly associated (P < 0.001) with sex in the two families. Models scanning simultaneously for the existence of two QTL or multiple QTL did not reveal any extra QTL.
Verification of SNP sex association and sex prediction
Sex association of 10 SNP markers (Additional file 3) selected from LG 13 was investigated using allele specific endpoint-genotyping assays (Figure 4; Additional file 3). The P-value thresholds (α = 0.05) after taking into account multiple testing (for the 10 SNP markers that were tested) according to the permutations and the Bonferroni correction tests were found to be 0.0054 and 0.0050 respectively. Hhi58665 (P = 3.05×10-19), Hhi10170 (P = 3.05×10-19), Hhi41238 (P = 2.45 × 10-16) and Hhi47769 (P = 8.96 × 10-17) showed the highest association with sex, over 97 per cent in the three test families (n = 10 ?:10 ? in Families 1 & 2; 9 ?:9 ? in Family 3; total 58). In each case the parents were a heterozygous ? and a homozygous ? for each marker. Furthermore when tested in 36 wild sourced broodstock from locations across the species native range (Additional file 4), the same four markers individually showed between 78 per cent and 89 per cent association with sex (P < 0.001), with SNP Hhi58665 showing the highest association (P = 6.39×10-7). These four markers were located within a 3.2 cM region of LG 13.

The combined prediction power of these four markers was tested on the 36 broodstock and 58 progeny using the JRip classifier, as the derived rules have straightforward interpretation. The combined prediction is based on two rules using three markers (Figure 5) and produced 97 per cent corrected classification with the 3 per cent of errors being phenotypic males predicted to be females. The male prediction precision is 1 (recall of 0.935) and the female prediction precision is 0.94 (recall of 1).
Within the 18 tested male broodstock, two had “female” genotypes for all four of these markers (one of the 58 progeny also had male phenotype but female genotype). One of these two broodstock had previously been crossed with four females and had produced only female offspring (between 3 and 14 individuals per family, total 27). The phenotypic sex of these 27 offspring was verified by post-mortem examination three years post-fertilisation.
Synteny searches
We selected the 59 markers within the 95 per cent confidence interval around the LOD score peak and mapped them onto the genomes of related species to identify syntenic regions. We performed this search against Danio rerio (zebrafish), Gadus morhua (Atlantic cod), Gasterosteus aculeatus (three-spined stickleback), Latimeria chalumnae (West Indian ocean coelacanth), Oreochromis niloticus (Nile tilapia), Oryzias latipes (medaka), Takifugu rubripes (tiger pufferfish) and Tetraodon nigroviridis (spotted green pufferfish) genomes. 33 markers had unique hits across at least five out of eight species (Additional file 5). G. aculeatus and O. niloticus show the highest level of synteny with H. hippoglossus and each other (Additional file 5). The order of the markers selected for SNP genotyping in the regions point toward one 3.2 Mb region embedding more than 60 annotated genes (See direct links to the Ensembl 68 in Additional file 5). No genes associated with sexual differentiation or determination were identified in this region.

Discussion
H. hippoglossus is a species of increasing commercial interest for cold-water marine aquaculture. However one of the main limitations to profitable culture of the species is the sexual dimorphism in age at maturation related to gender specific growth performance. To address this bottleneck, the current study demonstrates, for the first time in this species, that indirect monosex female production is possible for commercial H. hippoglossus aquaculture. While having strong commercial application, this research also had the fundamental aim to investigate the genomic regulation of sex determination in the species through state-of-the-art high-throughput sequencing methodologies.
RAD-tag sequencing has recently been used with a number of different fish species since the technology became available in 2008. One of the aims of the Baird et al study, which first validated the technique in fish, was to fine map QTLs in G. aculeatus. A number of different restriction-digest methodologies already existed, using high-throughput sequencers. However, what sets the RAD-tag methodology apart is the fact that it combines control over the fragments that result from the digestion with deep sequencing across individuals, making the identified SNP reproducible. This makes the RAD platform very efficient for constructing genetic maps and QTL studies.
In the present study, a genetic map of 5703 SNPs and 7 microsatellites spanning 1514 cM was constructed. To our knowledge this is the first dense genetic map incorporating SNPs in any flatfish species. The map has 24 linkage groups, corresponding to the number of chromosome pairs in H. hippoglossus. In a similar study, Amores et al. constructed a genetic map for Lepisosteus oculatus (spotted gar) consisting of 8406 SNPs. The above map was used to prove that L. oculatus diverged from teleosts before the Teleost genome duplication. Genetic maps of more than 4500 SNPs using RAD-seq were also constructed in O. mykiss and in D. rerio.
The high LOD (> 10), which was used to assign the genetic markers in linkage groups in our study, ensures that the map is of high quality. However, it must be acknowledged that even though the assignment of markers in linkage groups is robust, none of the available algorithms used for ordering markers provides an accurate positioning of closely spaced markers due to the relatively low number of meioses represented in our sample size. In a species like H. hippoglossus with no sequenced genome available, a genetic map is an invaluable tool for mapping any trait of interest in a QTL study. Apart from mapping QTL, the identified SNP of the genetic map can be used to construct a genomic relationship matrix, which can replace the relationship matrix inferred by pedigree for calculating breeding values. This would improve accuracy of estimated breeding values (EBV) under Best Linear Unbiased Prediction (BLUP) methodology in a breeding program. The improvement in accuracy is due to the fact that the genomic relationship matrix accounts for the random segregation of chromosome segments at meiosis between siblings.
In this study we associated mapped RAD-tag markers to sex determination. A major QTL involved in sex determination was identified in LG 13 in both families (LOD= 12.16 and 6.83 in Family B and C respectively). The location of the above QTL spans a region of around 22 cM. This region should contain one or more genes responsible for sex determination in H. hippoglossus. The reduced recombination in this region resulted in an almost flat likelihood surface for this region. Genome regions with reduced recombination are a common characteristic of sex chromosomes. In a similar study by Anderson et al. where the objective was to identify QTLs involved in sex determination in zebrafish using RAD-tag, a region in chromosome 4 spanning more than 20 cM showed reduced recombination. In general suppression of recombination keeps together genes (or alleles) with functions that are advantageous for one sex and avoids their transfer to the other sex chromosome, where they might have negative effects on the opposite sex.
Our data support the hypothesis that the H. hippoglossus has an XX/XY sex determination system. Among flatfish species, Paralichthys olivaceus (Bastard halibut) has also been shown to possess an XX/XY system, although temperature also influences sex ratio. On the other hand other closely related species, in which sex associated genetic markers have been identified, such as Hippoglossus stenolepis (Pacific halibut), Verasper variegates (spotted halibut), Scophthalmus maximus (turbot) and Cynoglossus semilaevis (half-smooth tongue sole) were all shown to have a ZZ/ZW sex determination system. Unusually in this group, C. semilaevis has differentiated W and Z chromosomes.
Validating the results of the QTL-Association Analysis is of the utmost importance. The fact that the sexassociated SNPs showed strong association when tested in a wider panel of three families and 36 wild broodstock provides clear evidence that those markers are in strong linkage disequilibrium with the sex-determining gene(s). Marker-assisted selection (MAS) could be conducted using these SNPs, providing a valuable tool towards more efficient production of all-female stocks for the aquaculture industry. In the current study it took four years from initiation of sex reversal treatment to completion of progeny testing for neomale identification with guaranteed all-female production from the following year. By employing MAS however it would be possible to confirm sex associated genotype from a non-destructive biopsy sample in hormonally-treated fish within 6-12 months of treatment, allowing neomales to be isolated and used from first maturation at three-four years posttreatment. SNPs Hhi58665, Hhi10170, Hhi41238 and Hhi47769 are the strongest candidates for MAS since they correctly assign sex in more than 97 per cent of the screened individuals. They span a narrow region of 3.2 cM. Genotyping a larger population for the SNPs in this region would allow fine mapping of the sex-determining locus. Other genetic factors involved in sex determination might also be involved.
The application of this technology will enable the industry to include a greater number of neomales from a wider genetic base to be included in future breeding programmes without the reduction in effective population size (Ne) associated with the use of a small number of neomales from these initial sex-reversed families. Limited examples exist of practical application of MAS in breeding programmes in aquaculture. A Y-specific DNA marker was used to assist in the development of monosex female culture in Oncorhynchus tshawytscha (Chinook salmon). More recently, MAS has been apply to a QTL for Infectious Pancreatic Necrosis Virus resistance in Salmo salar (Atlantic salmon): initially microsatellite markers were used, and more recently SNPs derived from RAD sequencing have been added.
Conclusions
Overall this work has demonstrated that all-female halibut production is commercially possible using indirect monosex production techniques. This in itself confirms that H. hippoglossus has an XX/XY sex determination system. RAD-tag sequencing produced 90,105 unique loci, and a single sex determination locus was mapped to LG 13. A further set of 4 markers that were present only or predominantly in DNA from male fish was isolated from two families and validated in a wider population screening, opening the possibility of MAS for sex in the species. Synteny analysis showed that DNA sequences containing H. hippoglossus sex-associated SNPs were consistently clustered in several other genomes, which provides a new focus for research into the sex determination mechanism in this species.
September 2013



