In this study, an exhaustive detection method was used to identify HGT genes in Litopenaeus vannamei. Homologous BLAST analysis was used initially to detect HGT genes with a view for identifying homologous genes of non-mating species from shrimp.
Initially, 65,582 gene segments were filtered out because there were no homologs detected among them. These sequences probably represent shrimp unique gene segments or non-coding DNA. In the second stage of homologous searching, more than 92 per cent of the gene segments, which were most similar to other arthropod sequence, were excluded. Then, only 965 HGT candidates were left. During this procedure, two classes of sequences were filtered out: gene segments that only showed homology to arthropods and those having higher BLAST similarity scores with arthropods than any other species.
However, several potential HGT events may be missed by this procedure, in that some truly HGT of Arthropoda may be considered as vertical inheritance. In the third homologous searching procedure, BLAST searches against the GenBank non-redundant protein database (nr) were implemented to extract sequences from an even larger spectrum of species. As small numbers of homologs are not sufficient for phylogenetic analysis and tree construction, we selected HGT candidates with more than 10 homologs in the nr database. Thus, after filtering genes in above procedures, only 722 HGT candidates remained for phylogenetic analysis (Figure 1).
Three kinds of phyloenetic analysis were performed on the remaining 722 HGT candidates after homology search (see Methods). Phylogenetic trees for each candidate HGT gene were constructed manually, and once a gene was nested within a donor clade and many other donor species formed basal branches, it was considered to be a HGT gene that had horizontally transferred from the donor to L. vannamei . Most of the HGT candidates showed higher similarity with Arthropoda-related species and grouped within a single clade. Furthermore, several candidate HGTs showed a phylogenetic topology that did not support a HGT event. Thus, as for those 722 HGT candidates, 693 of them were eliminated by the phylogenetic analysis, leaving only 29 genes that displayed rational HGT phylogenetic topology (Figure 1). They were subjected to comparative phylogenetic analysis by constructing three kinds of phylogenetic trees. Ultimately, well-refined maximum likelihood (ML) trees were used to represent the HGT events.
Among the 29 candidate HGT genes above after phylogenetic selection, there were 12 genes showed significant similarity to the best-hit sequences (E-values ranged from 2.00E-82 to 5.00E-36, and identity values ranged from 95.83 per cent to 100 per cent). Considering that many candidate HGT genes might be contaminants derived from RNA extraction, library construction or sequencing, these sequences needed to be excluded from the 29 candidate HGT genes. Previously, we acquired 42× coverage of shotgun reads generated from whole genome sequencing of L. vannamei. A 1.9 Gb draft map of the contigs was assembled and used to identify genuine shrimp gene segments. Ultimately, only 14 candidate HGT genes were homologous to shrimp genomic contigs (E-values ranged from 0 to 9.00E-21, and identity values ranged from 88.22 per cent to 100 per cent).
Thus, there are 15 genes were considered as non-shrimp genes and removed in this procedure. Some of HGT genes may be missed because of incompletely contigs assembly, but the 42× coverage of reads were full enough for identification of the existence of these gene segments. Generally, the sequenced reads tend to equally cover the assembly contigs, and eukaryotic contigs were expected to have different reads coverage to those contaminating contigs. Thus, with the help of SOAPaligner (http://soap.genomics.org.cn/soapaligner.html), we aligned all the reads to the 14 contigs which showed homologous with the 14 candidate HGT genes. The alignment results were compared with that of 1,000 randomly selected assembly contigs. It was found that the reads coverage of the 14 contigs (average coverage of 37.42) did not display any significant bias with that of 1,000 randomly selected contigs (average coverage of 38.25, Student’s t-test p < 0.05). Therefore, the 14 contigs seemed to possess eukaryotic characteristics rather than contaminates.
To avoid artificial mis-assembly of genomic contigs, PCR amplification was performed for these candidate HGT genes using shrimp genomic DNA as template. As expected, all 14 candidate HGT genes were successfully amplified. These amplified fragments were sequenced and showed nearly 100 per cent sequence identity with correspondent genomic contigs. Furthermore, in order to verify the 14 HGT genes were integrated into the genome of L. vannamei, PCR amplification was also performed on both sides of the 14 genomic contigs on which the 14 HGT genes located.
The two sides of the genome contigs, that did not show any similarity with the genome of the most probable donor, were most likely the eukaryotic sequences rather than HGT origin. BLAST results of the two sides of HGT fragments against nt database showed that most of them are similar to eukaryotic sequences (Additional file 1: Table S3). It indicates that the sides of these HGT fragments are eukaryotic portion, while the middle of the contigs is most probable HGT regions. Thus, we amplify the eukaryotic portion as long as possible to make sure the PCR products containing both eukaryotic and HGT regions. From the results, we can see that most of the edge products have been successfully amplified except for that of rpsN, which displayed a relatively weak amplification compared with the others. All the products were fully sequenced, and they were all identical to the correspondent regions of genome contigs.
For the control of PCR amplification, all the products of positive control has been successfully amplified, while the products of negative control were amplified with nothing (see Methods section), which support for accuracy of the amplification of our target products. From the results above, it is reasonable to confirm that the 14 HGT genes were integrated into shrimp genome. In Table 1, the assigned names of the 14 HGT genes were based on their annotated functions or the gene name registered in NCBI (acsf, rpsF, rpsN, exbB, mopB, tnpA, stat, rpc2, dhfr, cata, sdrp, omtp, ankp and deha).
Bacteria and fungi were the two predominating donors of HGT genes
The phylogenetic trees can be used to indicate HGT events and their directions. For 11 of the 14 HGT genes, the phylogenetic trees showed a phylogenetic topology of L. vannamei nesting with bacteria but far from other eukaryotes, which indicated a bacteria-to-L. vannamei HGT event (Table 1, Additional file 2: Figures S1, S2, S3, S4, S5, S6, S7, S8, S9, S10 and S11). The most probable donors of bacteria-to-L. vannamei HGT genes were Proteobacteria (five species) and Bacterioidetes (four species), and most of them were ecologically related to shrimp.
Desulfotomaculum kuznetsovii, Flavobacteriales bacterium, Robiginitalea biformata, Polaribacter irgensii, Gramella forsetii, and Vibrio harveyi are marine bacteria, while Escherichia coli and Bacteroides uniformis are gut bacteria. These results supported the view that HGT events generally occur between species inhabiting the same environment. The other two HGT genes were transferred from fungi to L. vannamei (ankp and deha, Table 1). Phylogenetic trees of ankp and deha showed that L. vannamei completely nested within the clade of fungi and far from bacteria and other eukaryotes, indicating a transfer of fungal origin (Additional file 2: Figures: S12, S13).
The best-hit species of these two HGT genes belong to the Ascomycota, which is the largest phylum of fungi. One of the two fungi donors, Grosmannia clavigera, was reported to be a symbiont of the mountain pine beetle. Few fungi-to-higher eukaryote HGT events have been detected in previous studies, whereas, some reports of fungi-to-higher eukaryote HGT genes indicated that they greatly improved the host’s phenotypes and evolution. Subsequent functional analysis indicated that these two fungi originated HGT genes may function in protein-protein interactions and electron transfer, respectively.
BLAST results of HGT genes against the nr database showed that the above 13 of the 14 HGT genes have no homologs among any other arthropods, which suggests that these HGT genes were transferred to L. vannamei or its ancestor after the speciation of Arthropoda. One exception was a HGT gene, omtp, which showed high similarity with sequences of other Arthropodarelated species (arthropods and nematodes) that were all nested within the group of bacteria while being phylogenetically far from other invertebrates and vertebrates (Table 1, Figure 2). Previous phylogenetic analysis clustered arthropods and nematodes in a clade of molting animals termed the Ecdysozoa, which indicated they are evolved from the same ancestor. Besides, omtp was considered to encode a catechol-O-methyltransferase (COMT), a protein family which has ever been found to be horizontally transferred from bacteria to eukaryotes before the separation of animals and fungi. This indicates that omtp was horizontally transferred earlier than the other HGT genes, and our phylogenetic tree of omtp suggested that it was transferred from a bacterial source to arthropods or its ancestors before the speciation of Ecdysozoa.
Four HGT genes contain introns
Structure analysis of HGT genes was used to investigate whether there is structural evolvement of the HGT genes between the donor and recipient, which may provide evidence of the HGT events. Seven HGT genes were derived from single EST or transcriptome sequences: tnpA (GI: 41059456), stat (GI: 57504717), rpc2 (GI: 57504772), dhfr (GI: 171573934), cata (GI: 259510079), sdrp (GI: 349908938), deha (GI: 171560878). When comparing HGT gene segments to the shrimp genome contigs, we found that four HGT genes (acsf, sdrp, ankp and deha) contained introns in the shrimp genome. ankp and deha are the two HGT genes derived from fungi and their homologous genes in fungal genomes also contain introns, whereas acsf and sdrp were transferred from bacteria, which were free of introns.
Surprisingly, it was found that the introns of genes from the donor fungi were different from those in the genome of shrimp, whereas the coding regions around the introns were highly conserved (E-values ranged from 8.00E-17 to 2.00E-13).
Besides, by alignment of shrimp genome sequencing reads to this gene using SOAPaligner, we found that the intron regions were in highly coverage of nearly 88X, which is significantly higher than the sequencing depth of the whole genome (42X). Furthermore, these two HGT genes are evenly covered at 76X, while the random selected 100 genes is covered at 48X. Through using SOAPsnp v1.03, three SNPs have been found on the contigs on which ankp located, and one of them has been detected locate on the intron ofankp. This indicated that the introns in two fungioriginated HGT genes may be highly repeated along with its surrounding sequences in shrimp genome. The shrimp acsf contains two introns (105 bp and 117 bp in length, respectively) and sdrp contains one intron (370 bp in length), but no introns were detected in the corresponding donor bacteria genome (D. kuznetsovii and B. phymatum). Therefore, it can be speculated that the introns might be integrated or changed in the HGT genes after horizontal transfer from the donor to L. vannamei.
Three large exogenous segments integrated into L. vannamei genome
When comparing the donor genome with the shrimp genome contigs, three pairs of relatively large segments showed significant similarity around the position of HGT genes (Figure 3). HGT gene tnpA, which encodes a transposase, is derived from a transposon of E. coli that comprises three transposase-encoding genes (tnpX, tnpR, tnpA) and non-coding regions. In addition to the whole transposon, there was also a kch gene (encoding a voltage-gated potassium channel) downstream of the transposon, and the whole segment (6,913 bp) was completely integrated into the genome of L. vannamei. Transposons generally contribute little or nothing to the host’s phenotype, but some horizontally transferred transposons have been identified to benefit the recipient’s genomic evolution. In addition to this large segment, there was one more gene, tonB, encoding a membrane spanning protein in the TonB-ExbB-ExbD complex, which was located downstream of the transposon in the donor genome. The other two genes (exbB and exbD, which encode ExbB and ExbD) were located far from tonB in the downstream. The exbB gene is one of the homologs of HGT gene exbB that was detected in this study (Table 1). It seems this horizontally transferred transposon might have served as a carrier for foreign genetic materials.
HGT gene rpsF was predicted to encode 30S ribosomal protein S6, which is involved in protein synthesis (Table 1). When compared with the most probable donor genome of F. bacterium (NZ_ABI01000003), it was found that rpsF was almost in full-length like that on the donor genome (Figure 3b). Furthermore, it was also completely mapped with shrimp genome contigs without any integrated introns. The corresponding regions of rpsF on the shrimp and bacterial genomes were highly conserved (E value of 2.00E-59 and identity of 87.5 per cent), which indicates that rpsF is an intron-less gene that may have transferred from bacteria to L. vannamei recently. Around rpsF, there is a large segment on the shrimp genome that is similar to the donor genome (Evalues ranged from 2.00E-75 to 1.00E-11, identity values ranged from 78.01 per cent to 92.31 per cent). This large segment, 4,655 bp in length, contains six genes on a corresponding donor bacterial genome (Figure 3). Besides, it is surprising to found that non-coding regions on this segment were not conserved, and also a predicted gene hypo2 was not conserved as well.
On donor bacterial genome, hypo2 is overlapped with both upstream hypo1 and downstream prsN. Homologous BLAST searching against the nt database detected a great many homologs (more than 100 matches) of this large segment, including hypo2. However, no evidence support hypo2 is actually a coding gene. If it is a gene prediction error, hypo2 may also a non-coding region. F. bacterium is just the best hit species of rpsF in GenBank database. It is known that there is a great deal of variation in genomes of bacteria. Therefore, the data of F. bacterium may be quite different from the true donor genome, which could lack hypo2. Thus, hypo2 may be missed in donor genome before the transfer of this large segment into shrimp genome. Whatever, the alignment results indicated that all these non-coding regions were absent from shrimp genome contigs. Similar results were also observed around mopB (Figure 3c).
The third horizontally transferred large DNA segment (4,118 bp) was around HGT gene mopB (Figure 3c). mopB is predicted to encode molybdopterin oxidoreductase and iron-sulfur binding subunit. Another molybdopterin oxidoreductase encoding gene, mopS, is located downstream of mopB and is also transferred to the shrimp genome. There is a region of low conservation (267 bp) upstream of mopB between the shrimp genome contig and the most probable donor genome. The alignment results indicated that most of the sequence (79.4 per cent) from this region was absent in the bacterial genome. Eukaryotic gene prediction software packages including AUGEST, Glimmer- HMM and FGENESH were used to predict exons in this shrimp genome contig, and all the prediction results indicated that the low conserved region was part of a complete exon but not an intron that had inserted into the gene. This suggested that shrimp might have redirected this gene to integrate an additional coding sequence into the gene.
HGT genes under strong negative selection pressure
In order to identify the completeness of these HGT genes, we surveyed them from the genomic contigs with the help of gene prediction software packages AUGEST and FGENESH. Even though some of contigs are short in length, we still detect some HGT genes (acsf, rpsF, exbB, mopB, rpsN, tnpA and dhfr) have complete gene structure, including transcription start site, introns, extrons, polyA and stop codons. Moreover, comparing with the genes on the most probable donor genome, there are not any frameshifts among these HGT genes. Therefore, these HGT genes are relatively complete on shrimp genomes.
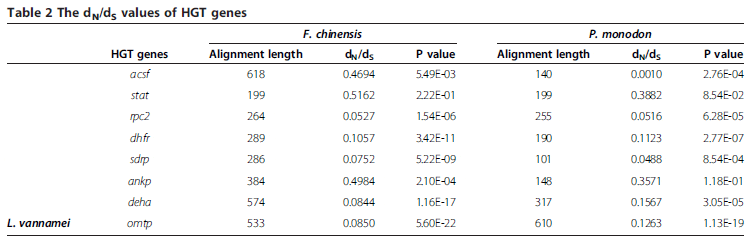
Then, more efforts were made on the detection of whether HGT genes are exposed to selective pressure after the shrimps diverged from their ancestor. Through comparing HGT genes against the transcriptome unigenes of Fenneropenaeus chinensis and Penaeus monodon (two kinds of shrimps that phylogenetically close with L. vannamei), we collected 16 pairs of orthologous sequences (Table 2). Then, KaKs_Calculator was used for calculating synonymous or nonsynonymous substitutions (dS, dN) among these pairs of genes. The statistical results showed that synonymous substitutions are relatively high in quantity (dS is around 0.7).
Except stat, almost all the ratios of the nonsynonymous substitutions per site to synonymous substitutions per site (dN/dS) among these pairs of HGT genes are significantly lower than 1 (Table 2), which indicated that most of these HGT genes are under strong negative selection (especially for rpc2, sdrp, deha and omtp, dN/dS values lower than 0.1). Similar results have been found in HGT genes of aphid, the dN/dS ratios of which are more than 0.3, which is larger than that of L. vannamei. Previous researches indicated that genes subjected to negative selection tend to maintain their functions. Therefore, most of the proteins encoded by these negative selected HGT genes appear to be functional.
Most HGT genes are predicted to be associated with energy metabolism and defense mechanism
Both TCA cycle and the electron-transport chain are predominant pathways of energy metabolism in the matrix of the mitochondria. The HGT gene acsf is predicted to Acetyl-CoA synthetase, which is involved in the metabolism of carbohydrate during the first step of energy generation in the tricarboxylic acid cycle (TCA cycle). Other four HGT genes (dhfr, sdrp, deha and mopB) are predicted as genes in electron transport (Figure 4). dhfr is an HGT gene annotated to encode dihydrofolate reductase which can use NADH as an electron donor to produce tetrahydrofolate for certain amino acids synthesis in 1-carbon transfer chemistry, and sdrp are predicted to encode NAD(H) or NADP(H) oxidoreductases. deha, predicted to be an HGT gene from fungi, encodes a cytochrome b5 reductase-like enzyme that catalyzes the reduction of cytochrome b5 using NADH as the electron donor. mopB is predicted to encode molybdopterin oxidoreductase which participates in transferring electrons to cytochrome c. Furthermore, a gene encoding cytochrome c, which contains a heme-binding domain for energy production and conversion, is located upstream of mopB and was transferred along with mopB (Figure 3). And at last, the energy-transducing protein ExbB, encoded by HGT gene exbB, is a part of TonB-ExbB-ExbD complex to form an energy transduction system. Therefore, according to the information of predicted functions, all these six HGT genes may associate with energy metabolism.
As for the six energy metabolism related HGT genes, it is of interest to find whether they are transferred from mitochondria in the distant past. It is well known that nuclear mitochondrial DNA (NUMTs) is commonly found to be transferred into nuclear genomes in various species [48]. However, in this study, none of six energy metabolism related HGT genes have been identified to show any similarity to the whole mitochondrial genome of L. vannamei (NCBI accession number of NC_009626.1). Therefore, unlike NUMTs, these HGT genes are not transferred from mitochondria in recent past, but transferred from nonmating organisms of shrimp in the long evolutionary history.
In Figure 4, the other three HGT genes are predicted to function in protein synthesis and interaction. rpsF and rpsN are predicted to encode two ribosomal protein subunits that are used for protein synthesis. ankp encodes a protein containing ankyrin repeats, which are used for mediating protein-protein interactions in diverse families of proteins. Interestingly, the spread of ankyrin repeats has been suggested to have occurred by HGT, and their occurrence in yeast excludes exon shuffling. This supported the HGT events of ankp, which maintained the stability of its gene structure.
According to the annotated functions of four of the HGT genes, they are associated with defense mechanisms. There are two antibiotic resistance genes (stat and cata) that are predicted to encode streptomycin 3′- adenylyltransferase and chloramphenicol acetyltransferase, respectively (Table 1). Both transferases are responsible for the streptomycin and chloramphenicol resistance and prevent them from binding to ribosomes. Accidently, streptomycin and chloramphenicol are specifically bind to 30S and 50S ribosomal subunit, respectively, which are predicted encoded by two other HGT genes, rpsF and rpsN (Table 1). Thus stat and cata may be involved in protection of protein synthesis. As the only bacteria-to -Arthropoda HGT gene in L. vannamei (Figure 2), omtp was annotated to encode O-methyltransferase, which is ubiquitously dispersed in various organisms and plays an important role in animal growth, development and defense. rpc2 is annotated to encode repressor protein C2, which is a gene involved in the SOS response. Generally, the SOS response is induced by various antibiotics, such as chloramphenicol, trimethoprim and streptomycin. The SOS response can promote HGT events of antibiotic resistance genes among bacteria. Above all, the proteins encoded by these four HGT genes are predicted as defense-related.
Gene expression of the HGT genes in differential developmental stages of shrimp
To detect whether HGT genes have effects on shrimp growth and development, we performed differential gene expression (DGE) analysis on these HGT genes, based on the transcriptomes of five developmental stages of shrimp larvae (see Methods). The number of reads aligned to the transcripts were calculated (Additional file 1: Table S4), and the expression level of each transcript was measured by FPKM values (fragments per kilobase of exon per million fragments mapped). High coverage of reads successfully supported for the following DGE analysis.
Nine HGT genes showed differential expression among five stages (Figure 5, Additional file 1: Table S5). stat displayed the highest expression level (FPKM values ranged from 1018.57 to 2910.86) at all five stages, while rpsF was expressed at the lowest (FPKM values ranged from 0 to 1.43). rpsF seems only expressed at the stage of zoea and mysis. Similar to rpsF, the other two fundamental DNA and protein synthesis related HGT genes (dhfr and ankp) displayed an expression pattern that gene expression level at the stage of zoea and mysis was generously higher than the other stages (Figure 5, Additional file 1: Table S4).
In addition to providing fundamental nucleic acid precursors and certain amino acids, dhfr also participates in energy metabolism. Three other energy metabolism-related HGT genes showed DGE, and two of them (acsf and sdrp) displayed similar expression pattern with dhfr. deha showed the reverse expression pattern, in that it showed lower expression at stages of zoea and mysis than at other stages (Figure 5). In addition, three defense-related HGT genes (rpc2, omtp and stat) were universally more highly expressed than other HGT genes (Figure 5, Additional file 1: Table S5), indicating that they may play an important role in early shrimp development.
Conclusions
Through sequence homology comparison, phylogenetic analysis and experimental verification, fourteen bacteria or fungi originated HGT genes were detected in L. vannamei. Spliceosomal-type introns were found to be inserted in four HGT genes, while non-coding regions of two large horizontally transferred segments were lacked on the shrimp genome. These structure alterations provide evidence that mature RNA may be the substrate for HGT events. Among 14 HGT genes, most of them were detected to be exposed to negative selection pressure and they appeared to be functional. Functions prediction annotated them to be associated with energy metabolism and defense of shrimp. Further studies should be taken to demonstrate the precise roles of these HGT genes.
October 2013


