Introduction
Characterization of microbiota in animal intestines has been central in advancing understanding of the relationship between host and microorganism. Microbial communities in humans and mice are estimated to comprise >1,000 taxa. While some microbes can be pathogenic to their hosts, other microbial symbionts are beneficial to the development and physiology of their host, playing roles in nutrient absorption, immune response, and epithelial development. Maintaining a balance in the population of intestinal bacteria is crucial to the health of the host. In turn, several host factors, such as diet, developmental stage and physiological condition have been identified to affect intestinal bacterial composition. The host, by its own gut immune system and gut environment (e.g. pH and bile acids), also shapes the composition of intestinal bacteria to maintain a relatively stable level of diversity.
In addition to understanding the host-microbiota relationship, characterization of intestinal bacteria will help identify bacteria with potential to become probiotics, which are increasingly used as an alternative means for preventive medicine in humans and animals. In aquatic animal farming, the concept of replacing antibiotic use with probiotics will help prevent environmental contamination and adverse health effects for consumers. Indeed, several studies have developed probiotic applications for aquaculture. This is no exception for Penaeus monodon (black tiger shrimp), which has been a commercially important marine crustacean for the past few decades in many Asian countries and Australia. With increasingly high shrimp consumption, the decline of wild harvests has forced domestication to become the major source of shrimp production. However, the farming industry has been deteriorating due to several factors, in particular the outbreak of disease.
As alternative means to address the disease outbreak problem, disease control using probiotics or maintaining healthy intestinal bacterial balance requires comprehensive understanding of bacterial diversity in the intestines of commonly farmed aquatic animals. Although there have been several studies on bacterial diversity in commonly cultured aquatic species animals from the wild such as P. merguiensis (banana shrimp), Salmo salar L (Atlantic salmon), Gadus morhua L (Atlantic cod), and Danio rerio (zebrafish), there are limited findings on the intestinal bacteria of P. monodon. It was only recently that studies of bacterial diversities in the intestines of P. monodon post-larval and juvenile stages were reported, yet there has been no report on the intestinal bacteria of wild-caught P. monodon, which feed on slow-moving benthic animals such as small crabs, shrimp, mollusks, and polychaetes. In contrast, domesticated shrimp in rearing facilities are fed with commercial feed pellets and reared in monoculture with higher stocking density. Comparing the differences in intestinal bacteria of wild-caught and domesticated shrimp can unveil changes in intestinal bacteria as shaped by aquaculture. These changes have implications for shrimp health and consequently, application of this knowledge may improve survival rates of P. monodon under domestication.
In this study, we examined bacterial populations associated with the intestines of wild-caught and domesticated adult black tiger shrimp using high-throughput next-generation pyrosequencing analysis in parallel with denaturing gradient gel electrophoresis (DGGE). Similarities and differences in intestinal bacteria between wild and domesticated shrimp provide initial evidence for a core bacterial community as well as probiotic candidates for P. monodon.
Results
Overview of 16S rRNA Pyrosequencing Analysis
Barcode pyrosequencing of 16S rRNA sequences was employed to determine bacterial populations in intestines of six individual adult shrimp from wild (WC, n = 3) and domesticated (DB, n = 3) habitats (Fig. 1). A total of 61,499 reads were obtained from pyrosequencing of the V3-4 regions of 16S rRNA genes (Table 1). The barcodes assigned the sequences to distinct libraries: WC1 (5,029 sequences), WC2 (9,969 reads), WC3 (11,532 reads), DB1 (2,961 reads), DB2 (3,190 reads) and DB3 (1,009 reads). Sequences were clustered into operational taxonomic units (OTUs) at 0.03 dissimilarity levels, in which each OTU represented a unique phylotype. The total numbers of OTUs ranged from 138 to 806, where the WC group exhibited higher variation in the number of OTUs. The OTUs were further classified to genus level using the RDP II Classifier. All six bacterial communities contained five phyla: Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Proteobacteria. DB1 from the DB group had the lowest number of bacteria genera (21, 43 and 28 genera for DB1, DB2 and DB3, respectively), whereas the highest number of bacteria genera was detected in WC2 from the WC group (39, 89, and 52 genera for WC1, WC2 and WC3, respectively).
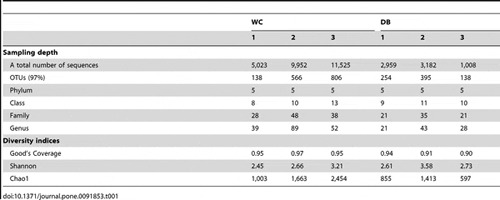
To estimate and compare bacterial diversity in each individual shrimp, bacterial diversity indices were calculated from OTUs of each library. Good’s coverage indices, used to estimate the percentage of total bacterial OTUs represented in a sample, were in the range of 0.90–0.97, suggesting the 16S rRNA results from each library represented the majority of bacteria in the shrimp intestines. Bacterial diversity estimated by the Shannon index varied from 2.45 to 3.21 in the WC group, and 2.61–3.58 in the DB group, suggesting a similar range of diversity between the two environments. Chao1 analysis for bacterial richness estimated a range of 597 to 2,454 phylotypes, higher than the actual observed OTUs in each library, indicating that the true bacterial richness in P. monodon intestines was underestimated. Additionally, rarefraction analysis was carried out to determine whether OTUs from each library had been adequately obtained from pyrosequencing analysis (Fig. 2). Consistent with the Chao1 analysis, rarefraction curves for the WC (Fig. 2A) and DB groups (Fig. 2B) did not plateau, suggesting that bacterial richness of adult P. monodon intestines were not yet determined. Additional sequence sampling will still be required to capture the true intestinal bacterial diversity of adult P. monodon. Even with the help from the next-generation pyrosequencing technique, a microbial ecology study to reveal a complete picture of bacterial communities is still very challenging. For instance, in soil bacteria community analyses, rarefraction curves have not been saturated, even when a range of 5,000 to 10,000 pyrosequencing reads were obtained.
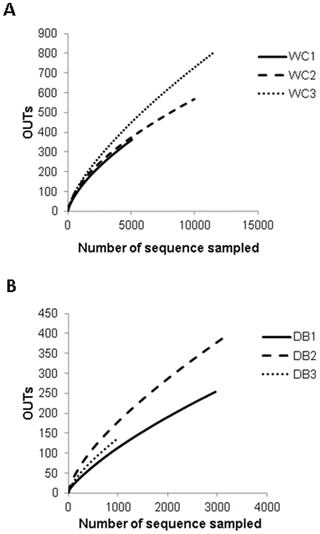
Operational taxonomic units (OTUs) were clustered based on 97 per cent sequence similarity for the (A) wild-caught group (WC1, WC2, and WC3) and (B) the domesticated group (DB1, DB2, and DB3). The number of sequences sampled represents the number of pyrosequencing reads.
Taxonomic Composition of Intestinal Bacteria in P. monodon from Pyrosequencing Data
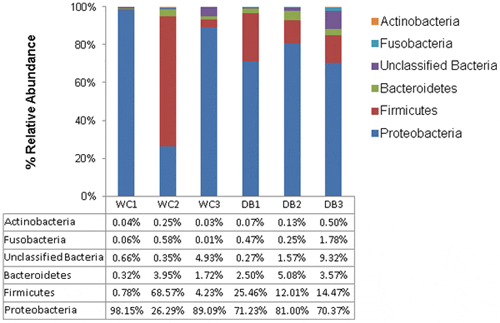
The five major phyla associated with P. monodon intestines were Actinobacteria, Fusobacteria, Bacteroides, Firmicutes and Proteobacteria (Fig. 3). Among them, Proteobacteria (>70 per cent of total sequences) dominated in all shrimp intestines, followed by Firmicutes, except for the WC2 library. WC2 library had 69 per cent of total sequences belonging to Firmicutes, and 26 per cent of total sequences were from Proteobacteria. Considering intra-group diversity, WC shrimp had more variation in phyla distribution whereas DB shrimp had relatively similar distributions, with Proteobacteria being the major phylum followed by Firmicutes (Fig. 3). To visualize bacterial composition in hierarchical orders, pie charts were constructed to represent bacterial abundance in intestines of WC and DB P. monodon (Fig. 4 and Fig. S1). Only phyla containing >100 total pyrosequencing reads were constructed into taxonomic nodes. Vibrio and Photobacterium were dominant in all six libraries, suggesting that these bacterial genera were well-adapted to conditions in P. monodon intestines and surrounding aquatic environments. Surprisingly, Lactobacillus was also present at high abundance in the WC2 library (Fig. S1A). Lactobacillus can be found in intestines of aquatic animals and marine environments, thus, the unexpectedly high abundance of Lactobacillus in the WC2 library could be possibly acquired from ingested food.
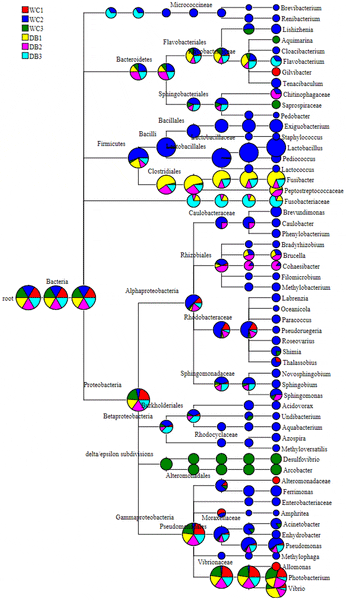
Pie charts of the hierarchical tree reflect relative abundance for each genus from each library (red represents DB1, blue represents DB2, green represents DB3, yellow represents WC1, pink represents WC2, and light blue represents WC3).
While the WC libraries were widely distributed in all taxonomic nodes, the DB libraries were mostly distributed in classes Gammaproteobacteria and Clostridia (Fig. S1). Interestingly, Fusibacter (class Clostridia) were found in higher proportions in the DB group than the WC group (Fig. S1B). Bacterial frequency and their classification are summarized in Table S1. Pyrosequencing reads that could not be classified at genus level were classified to the nearest taxonomic ranking (Table S1).
Evidence for Common Intestinal Bacterial Populations in P. monodon Adults
From the OTU (97 per cent similarity) distribution among the six libraries (Fig. 5), 1,541 OTUs were found to be unique within the WC libraries, and only 37 OTUs were shared within the group (2.4 per cent) (Fig. 5A). On the other hand, 689 unique OTUs were obtained within the domesticated libraries, and 18 OTUs were common within this group (2.6 per cent) (Fig. 5B), reflecting bacterial variations among individual WC and DB P. monodon. These bacteria could perhaps represent transient bacterial populations. Shared OTUs were assigned to the nearest species based on Blast analysis to determine common bacterial species (Fig. 5C). Interestingly, all 18 OTUs shared in the domesticated libraries were a subset of those 37 OTUs shared within the WC libraries, and these may be part of the core intestinal bacteria in P. monodon adults. The shared OTUs were classified to Proteobacteria, Firmicutes and Bacteroidetes. Phylum Proteobacteria contained the most shared OTUs among the six libraries, comprising Novosphingobium, Photobacterium, Pseudomonas, Sphingomonas, Undibacterium, and Vibrio (Fig. 5C). Among Firmicutes, a shared OTU was classified to Fusibacter paucivorans, whereas the other OTU could only be classified to phylum level. Only Cloacibacterium normanense from phylum Bacteroidetes was common across the WC and DB libraries.
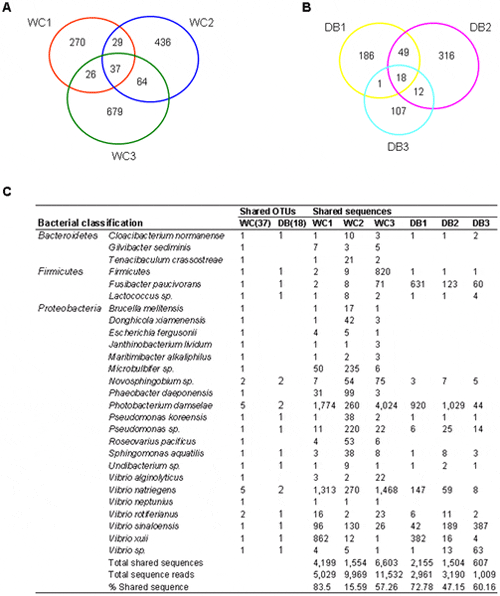
The Venn diagram represents the unique and shared OTUs at 97 per cent similarity level in libraries of (A) wild-caught group (WC1, WC2, and WC3) and (B) the domesticated group (DB1, DB2 and DB3). The shared reads were classified based on BLAST (C) with the following criteria; similarity of ≤97 per cent were classified to the nearest type strain, ≤90 per cent to the nearest genus, and ≤90 per cent to the nearest phylum.
On the other hand, there were distinct differences between the microbial structures of the WC and the DB shrimp. From 37 OTUs found from the WC libraries, the 19 OTUs absent in the domesticated included several bacteria previously found in marine sediment or marine organisms such as Gilvibacter sediminis, Microbulbifer sp., Donghicola xiamenensis, Tenacibaculum crassostreae, Maritimibacter alkaliphilus, Phaeobacter daeponensis, and Roseovarius pacificus. In addition, two Vibrio species were found only in the WC. Interestingly, an antibiotic-producing bacterium Janthinobacterium lividum was also found only in the WC.
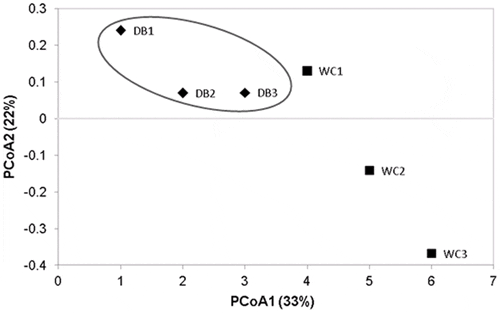
Principal coordinate analysis (PCoA) based on weighted-UniFrac distance showed that bacterial communities in the intestines of each shrimp in the WC were separated from one another, suggesting individual variation, whereas the DB group were clustered closer together (Fig. 6). The PCoA pattern suggested that bacterial populations in DB P. monodon showed more similarities within the group whereas the WC shrimp had higher variation. However, the statistical analysis of intestinal bacterial diversity did not show significant differences (P-value >0.05) between the WC and DB shrimp.
Analysis of Bacterial Diversity by Denaturing Gradient Gel Electrophoresis
The bacterial patterns in intestines of WC and DB shrimp were compared by denaturing gradient gel electrophoresis (DGGE) (Fig. 7). DGGE bands from each sample were selected for sequencing (Fig. 7A). The majority of DGGE bands from all six libraries were highly similar to bacteria from Vibrio and Photobacterium. One dominant DGGE band similar to Lactobacillus sp. (Fig. 7A, band B1) was only found in WC2. A sequence similar to Thalassomonas sp. (band C1) was also found in WC3. Sequences of DGGE bands similar to Fusibacter were found in DB1 and DB2. Consistent with the pyrosequencing result, cluster analysis of DGGE band profiles based on the Pearson distance matrix revealed that bacterial populations in WC shrimp showed higher individual variation than those in DB shrimp (Fig. 7B).
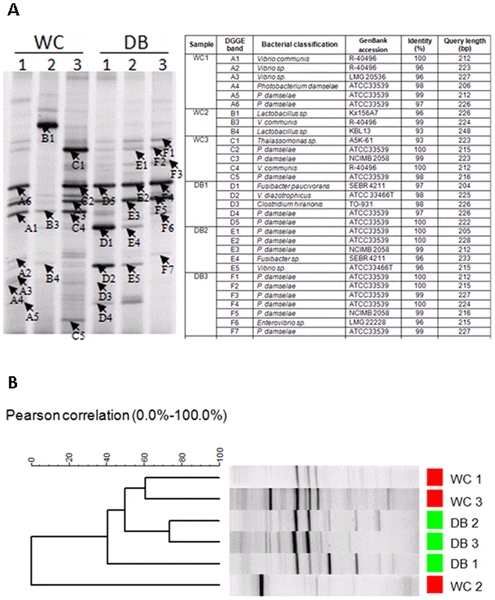
The bacteria profiles were from intestines of wild-caught shrimp (WC1, WC2, and WC3) and domesticated shrimp (DB1, DB2, and DB3). The selected DGGE bands were sequence to identify bacteria (A) and DGGE profiles were clustered using UPMGA based on Pearson correlation (B).
Discussion
The influence of intestinal bacteria on their hosts has been mostly elucidated in vertebrate hosts, with profound effects on host genes involved in nutrient absorption, mucosal modification and immune response. Some intestinal bacteria have been linked to the health status of their hosts. Due to the influence of intestinal microbiota on animal fitness, characterization of gut microbiota would be essential for farm animals. Here, we characterized bacterial populations associated with the intestines of P. monodon, an economically important shrimp species in Asia. To identify a core bacterial population, intestinal bacterial communities were determined in P. monodon obtained from wild and domesticated environments using a high-throughput pyrosequencing approach in parallel with DGGE analysis.
In this study, pyrosequencing analysis revealed some OTUs shared among P. monodon intestines from wild and domesticated environments. Although these OTUs represent only a small percentage of the total intestinal bacterial population, the dominant OTUs in each shrimp were commonly found in all six shrimp libraries. The closest relatives were classified to six genera from Proteobacteria (Vibrio, Photobacterium, Pseudomonas, Sphingomonas, Novosphingobiumcommon and Undibacterium), two genera from Firmicutes (Fusibacter and Lactobacillus), and one genus from Bacteroidetes (Cloacibacterium). Among the shared bacterial genera in this study, Vibrio, Photobacterium, Pseudomonas, Sphingomonas and Novosphingobium were found to be highly prevalent in the intestines collected from P. monodon post-larva and juveniles in our previous study. In particular, Vibrio and Photobacterium are commonly associated with marine habitats and many marine organisms such as crustaceans, oyster, and fish. Although the bacteria in these genera have been characterized as free-living or commensal in marine animal digestive tracts, the consistent detection of Vibrio, Photobacterium, Pseudomonas, Sphingomonas and Novosphingobium genera in this work and previous studies could imply that they are an indigenous bacterial population in P. monodon intestines. Some have been reported as pathogens in aquaculture, such as Vibrio harveyi, an opportunistic pathogen that causes infection in shrimp under conditions of high nutrient concentrations and high animal density in rearing environments. Fusibacter were commonly found in shrimp from both groups. Thus far, bacteria isolates belonging to the genus Fusibacter have been found to be strictly anaerobic and halotolerant bacteria. Interestingly, Lactobacillus was also found to be associated with P. monodon intestines. Members of Lactobacillus are indeed employed as probiotics in many animals, including some aquatic organisms. For instance, the group of Litopenaeus vannamei (Pacific white shrimp) fed with Lactobacillus plantarum supplemented diet shows a higher survival rate under pathogen exposure than the control diet group. The detection of Lactobacillus in P. monodon intestines in this report as well as our previous work also suggests that the bacterium could withstand P. monodon gut and aquatic environments. Hence, application of Lactobacillus as probiotics is a promising mean to enhance disease resistance in the P. monodon farming. Despite diverse intestinal bacteria members associated in P. monodon adults, the dominant bacteria detected in intestines of WC and DB P. monodon were Proteobacteria, followed by Firmicutes and Bacteroidetes. This observation is consistent with previous reports on intestinal bacteria in P. monodon post-larvae and juveniles. The prevalence of these phyla in animal intestines has been reported in aquatic organisms such as Fenneropenaeus chinensis (Chinese shrimp), P. merguiensis (banana prawn), Nephrops norvegicus (Norway lobster) and Danio rerio (zebrafish).
The similarity of bacterial communities among the wild and domesticated shrimp suggested the establishment of specific bacteria by selective pressures from the environment within the host gut. For instance, a germ-free mice model study reveals that mucosal environment influences selection and establishment of bacteria in the gastrointestinal tract. The mucosal immune system in a host’s digestive tract has been demonstrated to play important roles on maintaining commensal bacteria. Although understanding of P. monodon gut physiology remains elusive, expression of P. monodon immune-related genes upon pathogenic bacteria challenge provides evidence of a shrimp gut immune system. Furthermore, the observation of shared bacterial members is congruent with previous comparative studies of other wild and domesticated model animals. For instance, comparison of bacteria in intestines of D. Rerio (zebrafish) from wild and lab-reared environments showed similar bacterial members. Analyses of bacterial communities in wild and domesticated Mus musculus (mice), Drosophila melanogaster (fruit flies) or Hydra species (Hydra) reveal shared bacterial members regardless of individual variation, and similar bacteria compositions were observed at the phylum or class level in both wild and domesticated hosts. Finally, the similarities of intestinal bacteria in P. monodon with different life histories also imply that there has been long-term co-evolution between intestinal bacteria and host shrimp.
Although shared OTUs were observed among intestines of the six adult shrimps, there were still a vast number of unique OTUs, which reflect the high degree of bacterial variation among individual P. monodon, especially in wild-caught shrimp. In this study, bacterial profiles from the DGGE analysis were congruent with pyrosequencing results in that there was higher individual variation in intestinal bacterial populations among the wild-caught than the domesticated shrimp. As intestinal bacteria can be influenced by bacteria present in the surrounding aquatic habitats. Domesticated shrimp reared in the same water tank would therefore experience less fluctuation in bacterial composition in the surrounding water compared to wild habitats. Transiently persistent bacteria and those associated with ingested food could also inflate the observed individual bacterial variation. As wild P. monodon fed on smaller live animals, unlike domesticated shrimp fed mainly with dried feed pellets, the small live animals ingested by wild P. monodon might influence the number of intestinal bacteria genera, resulting in higher numbers of genera detected in the wild-caught P. monodon than the domesticated group. To further determine the effect of diets on intestinal bacteria, the characterization of bacterial compositions in P. monodon intestines is underway to compare differences between P. monodon feeding on live food (e.g. polychaete worms) to those fed with commercial feed pellets.
Most intestinal bacterial population analyses in penaeid shrimp have been characterized by both culture-dependent and culture-independent techniques based on Sanger sequencing of 16S rRNA genes and a recent study has applied the high-throughput 454 pyrosequencing platform to the analysis of intestinal bacterial diversity in P. monodon. The introduction of pyrosequencing technology has enabled a greater depth to bacterial population analysis in various environments; for instance, bacterial richness in the deep sea environment has been reported to be one or two orders of magnitude higher than those obtained under the traditional Sanger based approaches. Despite its advantages, even pyrosequencing may not uncover the entire range of bacterial diversity. Although the reading length obtained from pyrosequencing technology has been continuously improving, the full length of 16S rRNA (~1.5 kb) has not yet been achieved with current 454 pyrosequencing capability. Primer selection and their target regions influence bacterial diversity analyses and indeed the detection of phyla Verrucomicrobia, Planctomycetes and Chlamydiae is highly variable with 16S rRNA primer sets. We chose primers target variable regions 3 and 4 of the 16S rRNA gene, which may not be effective in detecting the presence Verrucomicrobia and Planctomycetes. Nonetheless, these bacteria phyla were not found in high abundance in P. monodon intestines in a previous study using full-length 16S rRNA clone library approach. While pyrosequencing analysis provided more comprehensive view of bacterial diversity in a community, the DGGE analysis in parallel provided an overview of structures of dominant bacterial groups, which can be used for comparison of bacterial patterns.
Our work reports intestinal bacterial composition in wild-caught and domesticated P. monodon adults by using the 454-pyrosequencing approach and bacterial patterns were also compared by DGGE analysis. We provided evidence of common intestinal bacteria in P. monodon intestines, and suggest that the intestine may be viewed as a selective environment that allows only certain bacterial taxa to persist. Characterization of the bacterial composition in P. monodon intestines is fundamental to understanding of the beneficial effects of bacteria and the importance of host-microbe interaction in a non-model organism.
Conclusion
Recent studies have reported the importance of balanced gut microbiota to the health of animal hosts. Hence, the main purposes of this study were to utilize a culture-independent high-throughput pyrosequencing technique to extend our knowledge concerning the breadth of bacterial diversity in intestines of adult P. monodon, and to determine the composition of bacterial communities in P. monodon from the wild and those under domestication. The bacterial profiles showed similar dominant genera in wild-caught and domesticated shrimp, suggesting the occurrence of a resident bacterial population in P. monodon. We identified shared bacterial members associated with adult shrimp intestines from wild and domesticated environments. This work provides evidence that host intestinal conditions exert stronger selective pressure for bacterial community establishment in P. monodon than rearing environments.
April 2014

