Edwardsiella tarda, a member of the family Enterobacteriaceae, has been isolated from a variety of animals including fish and mammals. In pathology, this bacterium is a known causative agent of a fish disease (e.g. gangrene and septicemia) named edwardsiellosis. Ever since the first report of edwardsiellosis in 1959, the mass mortality of fish caused by this bacterium has been a serious issue in aquaculture. E. tarda can infect a variety of fish species, including Japanese eel (Anguilla japonica), European eel (Anguilla anguilla), Japanese flounder (Paralichthys olivaceus), turbot (Scophthalmus maximus), yellowtail (Seriola quinqueradiata), red sea bream (Pagrus major), channel catfish (Ictalurus punctatus), and tilapia (Oreochromis mossambicus). E. tarda also causes diarrhea in humans.
The type and virulence of the E. tarda strains have been examined by serological analysis and infection test, respectively. The isolates from Japanese eel, Japanese flounder and eel pond, were classified into four serotypes (A, B, C, and D) by the O-agglutination test. The E. tarda that are highly virulent to fish are serotype A strains, but these strains do not always share the same biological traits. In particular, atypical serotype A strains of E. tarda isolated from red sea bream and yellowtail were non-motile, unlike the more typical serotype A strains. To investigate the virulence of E. tarda in fish, the infection test was performed using both the Japanese flounder and red sea bream as hosts. While all the serotype A strains of E. tarda are, in principle, virulent to Japanese flounder, the atypical strains were reported to be virulent only in red sea bream.
Regarding the genomic data of E. tarda, a complete genome sequence of the turbot pathogenic strain EIB202, was reported in 2009 and strain FL6-60 was sequenced in 2011. The genome sequence of the human pathogenic strain ATCC23685 was also determined and annotated, but the sequence is still fragmented. In addition, the complete genome sequence of Edwardsiella ictaluri, a close relative of E. tarda and causative agent of enteric septicemia in catfish, is currently available. A recent whole genome comparison of multiple E. tarda strains showed that E. tarda genotypes were broadly clustered into two groups, EdwGI and EdwGII, which consisted of strains that were isolated mainly from fish and human, respectively. EdwGI represents a genotype of fish pathogens in the Edwardsiella lineage and the genes of virulence factors such as type III secretion system (T3SS), type VI secretion system (T6SS), hemolysin, flagellin, adhesin, invasin, and fimbriae have been identified in strains from this group.
The relationships between the EdwGI and EdwGII genotypes and the A–D serotypes are not fully understood. Serotype A strains are virulent to fish, indicating that these strains are evolutionarily closely related to the EdwGI genotype. On the other hand, two unique DNA sequences from atypical serotype A strains have been detected. These DNA sequences were found to encode a novel T6SS and the type V secretion system (T5SS). Thus, there is a possibility that the virulence mechanism of serotype A/EwdGI E. tarda may differ between the typical and atypical strains, consistent with the reported host specificity in the infection test. In this study, we sequenced the genomes of four serotype E. tarda isolates (serotypes AD) from aquaculture fishes or environmental water, and performed comparative analyses of the structure of the genomes and their virulence-related gene repertoire using the reference genome sequences such as those of EIB202 and ATCC23685. We demonstrated that fishpathogenic and environmental E. tarda were clearly distinguishable at the sequence and gene repertoire level, and found that a single genotype proposed previously for fish-pathogenic strains could be further classified into two genotypes, typical and atypical. Strikingly, we report that an atypical strain of E. tarda has a pathogenicity island that is homologous to the pathogenicity islands of virulent Escherichia coli strains, which are causative agents of outbreaks of human foodborne illness.
Results and discussion
Genome assembly
The complete genome sequences of the eight E. tarda strains, E22, NUF806, FPC503, SU100, SU117, SU138, SU244, and ATCC23685, ranged in length from 3.63 to 3.96 Mb (Table 2). The estimated genome sizes were similar to those of the previously determined strains (EIB202: 3,760,463 bp; FL6-60: 3,684,607 bp) and E. ictaluri (93–146: 3,812,315 bp). The GC content ranged from 57.2 per cent to 59.8 per cent. The GC content of the three fishpathogenic strains (NUF806, E22 and FPC503) was close to that of EIB202 (59.7 per cent) and FL6-60 (59.8 per cent) and around 2 per cent higher than the GC content of the other four strains (SU100, SU117, SU138, and SU244). The four strains with the lower GC content are the environmental strains that were isolated either from pond or healthy eel gut, and their GC content was similar to that of E. ictaluri (57.44 per cent). Thus we found that the fish-pathogenic and environmental strains of E. tarda were distinct from each other at the GC level.
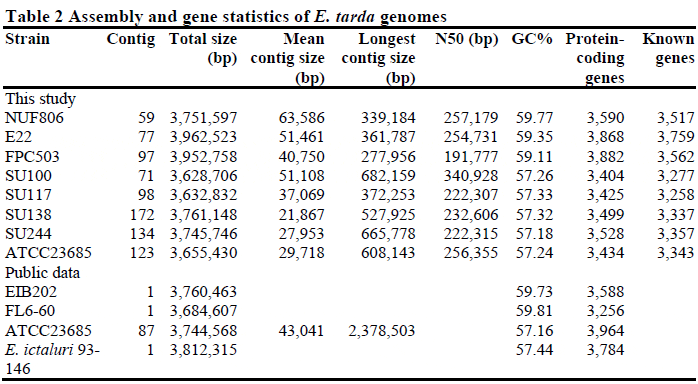
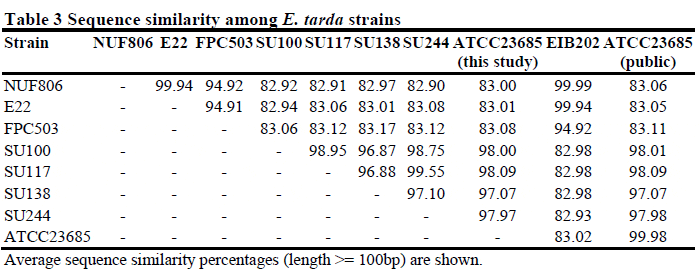
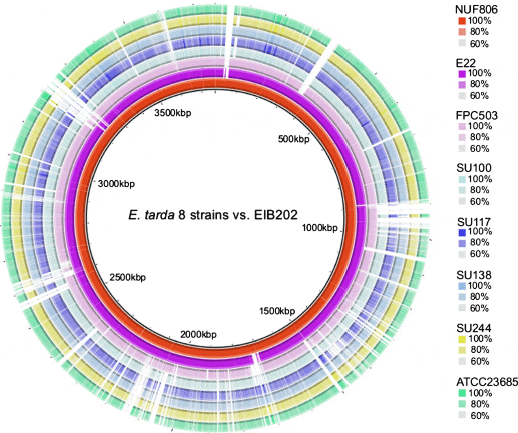
To evaluate the assembly statistics, we resequenced the public E. tarda strain ATCC23685 in parallel with the other seven E. tarda strains, and compared the data (Additional file 3: Figure S3). For ATCC23685, we obtained 123 contigs consisting of 3,655,430 bp by de novo assembly; the public sequence had 87 contigs consisting of 3,744,568 bp. A total of 3,605,608 bp (98.6 per cent) of the 3,655,430 bp mapped to the public scaffold sequence, and more than 99.9 per cent of mapped nucleotides were identical. We compared the average identity of all the sequenced genomes among all the strains of this study, and found that the fish-pathogenic and environmental strains were clearly different from each other at the sequence similarity level (Table 3). The nucleotide sequence of the FPC503 (from red sea bream) was similar to the NUF806 (flounder) and E22 (eel) sequences, but differed by about 5 per cent. Using the genome sequence of strain EIB202 as the reference, we compared the genomic structure among the eight strains by contig mapping (Figure 1). We found that the EIB202 genome was covered almost entirely by the contigs of NUF806 and E22, but some loci in the EIB202 genome were absent in the other six strains. Indeed, the EIB202, NUF806 and E22 genomes are highly similar at the sequence level (Table 3), indicating that, of the eight strains, these three strains are the most closely related.
Gene prediction and validation
We detected 3400–3900 ORFs in the sequenced E. tarda strains (Table 2). Of these predicted genes, an average of 96 per cent (3258–3759 genes, excluding ATCC23685) matched known sequences. For ATCC23685, we predicted a smaller number of ORFs (3434 genes) than was predicted in the public reference data (3964 genes); 3276 of the genes were common to both sets of data as predicted by BLASTP. One reason why the gene numbers are different between the two sets of ATCC23685 sequence data might be inaccuracy in genome assembly. The ATCC23685 sequence obtained in this study has more contigs (123 contigs) and a shorter average length than the reference sequence (Table 2), implying that genes split by gaps between contigs have been missed by the gene-finding software. Another feasible reason may be that the reference data are of low quality. We checked the reference gene annotations and found that 302 genes have incorrect lengths (indivisible by three), suggesting that some of the reference genes are either pseudogenes or have been overestimated by falsepositives (Additional file 4: Figure S4). Using mutual TBLASTN to query the protein sequences against the contig sequences, we were able to find almost all of the missing genes in each ATCC23685 sequence. Finally, we confirmed that a total of 3426 (99.8 per cent) genes in our sequence were also present in the reference sequence, and 3934 (99.2 per cent) genes in the reference sequence were present in our ATCC23685 sequence. Thus, we concluded that the genome data of the E. tarda strains of this study covered more than 99 per cent of protein-coding loci and are accurate enough to be further compared.
Gene comparison
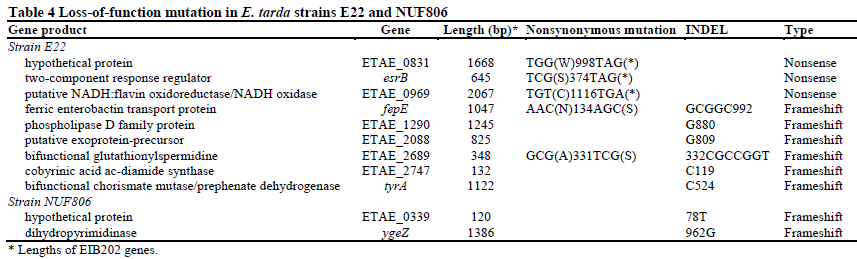
To detect genetic differences between the E. tarda strains, we focused first on SNPs and INDELs. We mapped the NUF86 and E22 reads to the turbot pathogen strain EIB202 genome, because we had found that the sequences were highly similar to each other (Figure 1 and Table 3). We predicted a total of 79 SNPs or INDELs between NUF806 and EIB202, and 355 between E22 and EIB202 (Additional file 5: Table S1). Although most of the detected SNPs or INDELs were located in non-coding regions, 40 and 242 SNP/INDEL candidates were in the coding regions in NUF806 and E22, respectively. In this study, we focused on nonsense or frameshift mutations in protein-coding genes (Table 4), because such mutations are more likely to result in loss of function of the proteins that they encode. We found nine genes in E22 and only two genes in NUF806 that contained loss-of-function mutations. In particular, E22 had a nonsense mutation in the esrB of T3SS, which is involved in the virulence of E. tarda. Because the E22 strain has been attenuated during cultivation, a few mutations may have occurred in a short period. We propose, therefore, that the mutation in esrB may be responsible for the attenuation of this strain.
We performed an all-vs-all BLASTP using the gene sets of eleven Edwardsiella strains (NUF806, E22, FPC503, SU100, SU117, SU138, SU244, and public E. tarda strains EIB202, FL60, ATCC23685, and E. ictaluri 93–146). We found that at least 2422 genes were conserved among all the strains, and 4147 genes were polymorphic, that is, each gene was absent from one or more of the eleven strains. We converted the polymorphism (presence/absence) of genes into a distance matrix and conducted cluster analysis. The dendrogram that we obtained was congruent with the molecular phylogenetic trees (Figure 2), suggesting that gene gain/loss events reflect the evolutionary scenario of the Edwardsiella lineage. In particular, the gene catalogues of the fish pathogen and non-pathogen strains were clearly distinct from each other, consistent with the previous study. In this topology, E. ictaluri was positioned between pathogenic and environmental E. tarda, suggesting that the classification and nomenclature of Edwardsiella species may need to be reconsidered. Moreover, all the serotype A strains, the typical (NUF806 and E22) and the atypical (FPC503), were classified into a single genotype EdwGI; the other serotype strains were clustered with ATCC23685, which has an EdwGII genotype (Figure 2B). It should be noted that FPC503 constituted a different clade from that of the typical serotype A strains, suggesting that the EdwGI group may be composed of two subgroups.
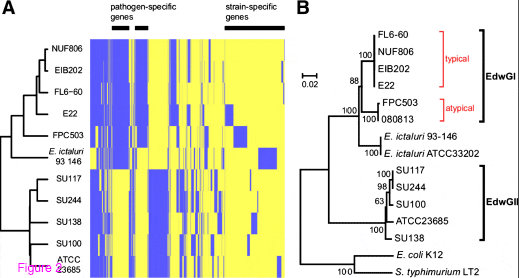
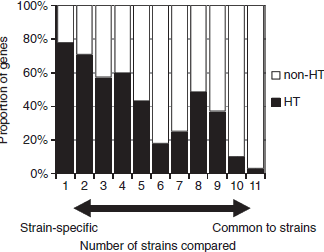
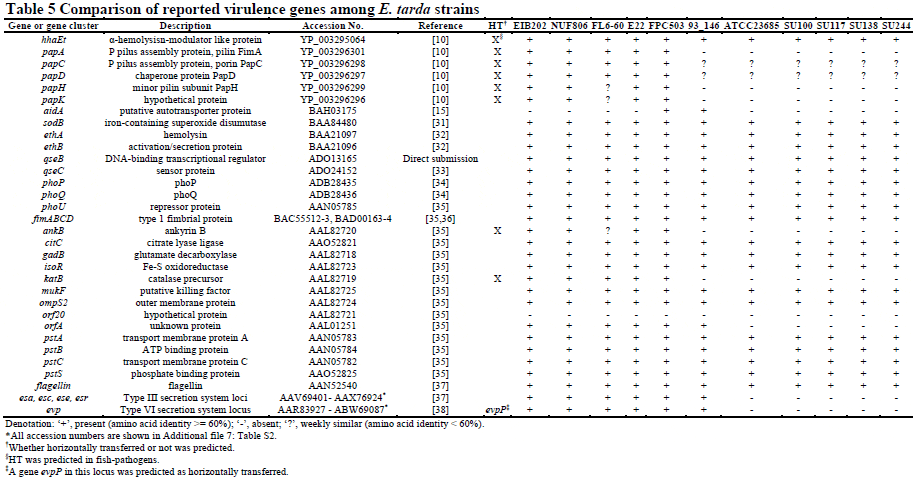
To investigate the origin of the polymorphic genes among E. tarda strains, we conducted a horizontal gene transfer analysis (Figure 3). We found that most of the strain-specific genes tended to be horizontally transferred (HT), while most of the common genes were non-HT genes. Interestingly, the proportion of HT genes dropped around six strains as shown in Figure 3. This result can be explained by our experimental design: six fish-pathogens (NUF806, E22, FPC503, EIB202, FL6-60, and E. ictaluri 93–146) and five non-fishpathogens (SU100, SU117, SU138, SU244, and ATCC23685), which corresponded to two phylogenetically distinct clades (as described above), were used in the study. Thus, the observed paucity of HT genes around six strains probably reflects clade-specific loss events of ancestral genes. One may speculate that the HT genes detected in this study may be artifacts due to DNA contamination in sequencing. However, we note that the HT genes common to E. tarda strains were distributed preferentially to either of the two clades (Additional file 6: Figure S5), likely reflecting the gene gain events in each lineage. In addition, many (121/323) of strain-specific HT genes annotated were mobile element genes, such as phage-, plasmid, or transposon-related ones, which is unexplainable by DNA contamination. The presence/absence of virulence genes in E. tarda is summarized in Table 5 (Additional file 7: Table S2). Fish-pathogenic strains have two secretion system genes (T3SS and T6SS) and pilus assembly genes. We predicted that the T3SS and T6SS genes are both non-HT genes, while the pilus assembly genes are HT genes. We concluded that the T3SS and T6SS genes originated in an ancestral Edwardsiella lineage and were subsequently lost in non-pathogenic E. tarda. However, here we noted that a gene in the T6SS locus, evpP, was predicted as being an HT gene. The evpP gene is located at the end of the T6SS locus; therefore, it may have been added to the locus after the divergence of pathogenic- and non-pathogenic E. tarda. Particularly, it has been shown that deletion of evpP in E. tarda significantly decreased the virulence of the pathogens in fish. Here, we propose that the ancestral T6SS of the Edwardsiella lineage was not originally involved in pathogenesis and that the subsequent acquisition of evpP contributed to the virulence of E. tarda. We also compared the genes related to biosynthesis of lipopolysaccharides as O-antigens among the E. tarda strains, and found polymorphisms related to the presence/absence of rfb homologs (Additional file 8: Table S3), possibly due to horizontal transfer. The serotype A strains (NUF806, E22 and FPC503) share all the genes reported in E. tarda EIB202, which is characteristic of genotype EdwGI. Non-pathogenic strains (SU100, SU117, SU138 and SU244) are different from the serotype A strains and also from each other. This presence/absence of rfb polymorphism might explain why non-pathogenic strains have different serotypes (B to D).
NUF806-specific genes
Among the eight sequenced strains in this study, we observed that NUF806 and EIB202 were the closest at the genome sequence level; almost all the genes were common to both strains. However, unlike EIB202, NUF806 lacked plasmid-encoding genes, namely, the type IV secretion system (T4SS) that is involved in conjugative transfer of plasmid, and the drugresistance genes against streptomycin and chloramphenicol. Therefore, NUF806 may be sensitive to these antibiotics. Because NUF806 and EIB202 are flounder pathogens with similar virulence, this finding suggested that the plasmid-encoding genes are not essential for pathogenesis in flounder.
E22-specific genes
Among the eight strains in this study, E22 is the second closest strain to EIB202. Although there were no major differences in the gene sets of the two strains, we found that loss-offunction mutations had occurred in some of the genes (Table 4). On the other hand, we found that E22 had plasmid-related genes which were almost identical to corresponding genes in a conjugative plasmid (pRA1) isolated from a fish-pathogenic bacterium, Aeromonas hydrophila. The plasmid genes were encoded in four contigs with a total length of 140 kb, which covered more than 90 per cent of the pRA1 genome (Additional file 9: Figure S6). Because the gene that encodes RepA (plasmid replication protein) and conjugative transfer genes were included in the region, the contigs probably constitute an intact plasmid which is not integrated into the E22 chromosome. The plasmid of E22 also carries drug-resistance genes, tetRA for tetracycline, sul2 for sulfonamides, and hipAB for beta-lactams. Previously, it was reported that many of the pathogenic E. tarda strains isolated from eel were resistant to tetracycline and sulfamonomethoxine, probably because of continued drug treatment in eel ponds. The previous study had demonstrated that such drug-resistance markers may be located on an 81-kb conjugative plasmid. We propose that the longer E22 plasmid is evolutionarily related to the previously reported 81-kb conjugative plasmid, and that these may share a common ancestor with the plasmids isolated from A. hydrophila.
FPC503-specific genes
We found that FPC503 had genes of the novel T3SS and T6SS which are not present in the other E. tarda strains in this study. These genes were predictable in strain 080813 which is a close relative of FPC503 (Figure 2), although the contigs of 080813 are still fragmented (T3SS, [GenBank:AFJH01000035]; T6SS, [GenBank:AFJH01000029]). Therefore, the second T3SS and T6SS were considered to be a common feature of the atypical E. tarda, which is distinct from the typical strains. At the sequence level, the second T3SS was similar to the T3SS of E. coli, and the T6SS was similar to the T6SSs in other enterobacteria, Enterobacter and Pantoea. To examine the locus structures in detail, we sequenced the genome of FPC503 using longer-read 454 pyrosequencing. De novo assembly produced a single contig for the T3SS locus, and two contigs for the T6SS which were further joined into a single contig by PCR-based genome walking. Both contigs contained, at either end, the genes that were present in the E. tarda EIB202 chromosome, implying that these contigs were derived from the FPC503 chromosome and not from the plasmids. We observed that homologs of intimin and Tir (translocated intimin receptor) were encoded in the T3SS cluster. These genes (eae and tir) are known to be important elements in a pathogenicity island of enteropathogenic and enterohemorrhagic E. coli strains, namely the locus of enterocyte effacement (LEE). Strikingly, when we compared the gene content and order between the FPC503 T3SS cluster and the E. coli LEEs, we found that they were well conserved (Figure 4A and Additional file 10: Figure S7A). Indeed, 29 out of 42 genes in enteropathogenic E. coli (and 28 out of 40 genes in enterohemorrhagic E. coli) were identified in the FPC503 T3SS locus, and the observed differences in the gene order were explainable by assuming a few recombination events. Furthermore, we observed microsynteny in each of the five major operons (LEE1, LEE2, LEE3, LEE4, and TIR), which constitute LEE. Thus, we concluded that FPC503 had a LEE-like pathogenicity island that we named Et-LEE (E. tarda LEE). For the second T6SS, which we termed Et-T6SS2, we also observed a high synteny to a T6SS cluster in P. ananatis (Figure 4B). In particular, we found a homolog of vgrG that encodes an effector protein of T6SS. As reported in other enterobacterial genomes, this gene is closely located to hcp, which was identified previously in E. tarda, suggesting that these genes may function as essential components of the Et-T6SS2 in FPC503. In the genome assembly of FPC503, we found another contigs that were similar to the Et-T6SS2 locus (Additional file 10: Figure S7B), implying that this locus was duplicated in FPC503.
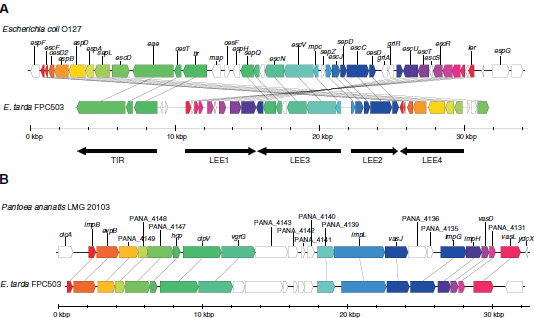
It is known that pathogenicity-related genes often flow among species by horizontal gene transfer . Using a Markov model method, we predicted that Et-LEE was extrinsic to FPC503 through recent horizontal transfer. The T6SS locus was not significantly predicted by the method, but the genes may possibly be of the horizontal origin because the gene sequences were highly similar to the corresponding genes in Pantoea (average amino acid identity = 80 per cent) and no orthologs were present in other E. tarda strains. A difference between E. coli LEE and Et-LEE is their locations in the genomes: E. coli LEE was generally inserted next to a tRNA locus, but no tRNA locus was found close to Et-LEE. In addition, no transposable element related genes were detected near the Et-LEE, except for a member of the transposase IS3/IS911 family. Therefore, we proposed that Et-LEE may either have lost mobility after integration or have been inserted in a different manner than E. coli LEE.
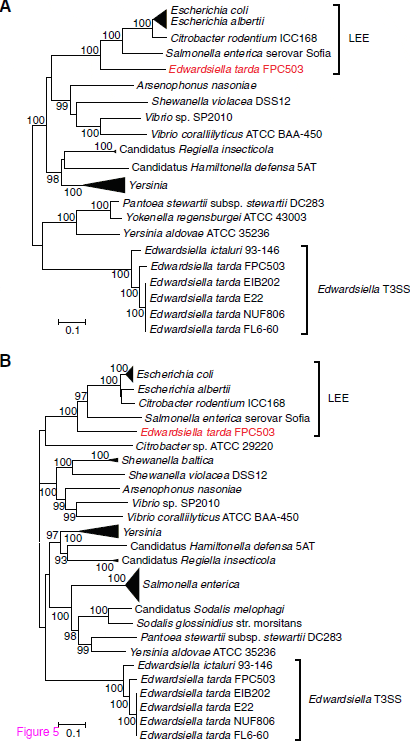
Our result raises a further question about why FPC503 acquired and retained Et-LEE. Since, in E. coli, the secreted Tir and intimin proteins encoded in LEE function in adhesion to intestinal epithelial cells, Et-LEE may also play a role in the intimate attachment of the pathogen to fish intestinal cell. We should keep in mind that FPC503 is a non-motile strain (Table 1), a trait that is disadvantageous for infection to host cells. Thus, a plausible explanation for the acquisition of Et-LEE by FPC503 may be that Et-LEE can compensate for its non-motility: when FPC503 is carried close to the host intestinal cells, it can fix tightly and effectively colonize its host by using Et-LEE. The origin of LEE in enterobacteria is also an unanswered question. LEE has been reported in pathogenic E. coli, in a mouse-pathogen Citrobacter rodentium, and in Salmonella enterica, but, until now, it has not been reported in fish pathogens. The current study has shown that the E. tarda strain that infects red sea bream may have also acquired Et-LEE by horizontal transfer, meaning that the donor species of LEE was not E. tarda. Molecular phylogenetic analysis indicated that all the Et- LEE genes examined were significantly close to the LEEs of E. coli, C. rodentium and S. enterica (Figure 5 and Additional file 11: Figure S8), suggesting that Et-LEE may be an appropriate outgroup of these LEEs. The sequencing of other E. tarda strains that harbor Et LEE (e.g. strain 080813) may fill a missing link in the evolution of pathogenesis associated with LEE in enterobacteria.
Conclusions
In this study, we determined the genome sequences of eight strains of E. tarda using nextgeneration sequencing technology. The GC content, hierarchical clustering based on gene repertoire, and phylogenetic tree, all clearly showed differences between the fish-pathogenic and environmental E. tarda genome sequences. By comparing the genomes, we identified polymorphisms that were responsible for serotypes and for the pathogenesis of E. tarda. We found that O-antigen related genes were different among each of the serotype strains, and that fish-pathogenic E. tarda was characterized by having two types of secretion systems (T3SS and T6SS) and pilus assembly genes. We predicted that the lineage- and species-specific genes may have originated by horizontal transfer, perhaps providing E. tarda with important traits that could be used as strain-dependent drug targets in aquaculture. Importantly, in this study, we found that the E. tarda strain that was isolated from red sea bream had T3SS (Et- LEE) and T6SS (Et-T6SS2) genes that were of horizontal origin from foreign organisms. This observation suggests that the previously proposed E. tarda genotype EdwGI could be divided into two sub-genotypes, a typical one and an Et-LEE/T6SS2-bearing (atypical) one. This is the first report that a fish pathogen possesses LEE, which is known in zoonotic pathogenic enterobacteria. This finding may provide a clue to the origin of the LEE pathogenicity island. Our results suggest that gene flow beyond species has a wide influence in the pathogenesis of enterobacteria.
January 2014

